概要
FAAH遺伝子欠損マウスの脊椎のメタボロームデータ(Wild type,Knock outマウスそれぞれ6サンプル)を利用し,xcmsを用いた質量分析データ処理の流れを説明する.
- データの説明
- Fatty acid amide hydrolase (FAAH) : 脂肪酸アミドのアミド結合を切って脂肪酸とアミンに分解する酵素
- アナンダミド(脳内麻薬物質の一つ)→エタノールアミン+アラキドン酸
- FAAH阻害薬は創薬ターゲットになっているが、様々な重篤な副作用が報告されている
XCMSのドキュメント「LCMS data preprocessing and analysis with xcms」の(かなりの)簡略版
ファイルの読み込み
faahKOパッケージには,ポジティブイオンモードのNetCDFファイルが含まれており, まずはじめにこのファイルを読み込む.
library(xcms)
library(faahKO)
## load raw data
cdfs <- dir(system.file("cdf", package = "faahKO"), full.names = TRUE, recursive = TRUE)
cdfs## [1] "/usr/local/lib/R/site-library/faahKO/cdf/KO/ko15.CDF"
## [2] "/usr/local/lib/R/site-library/faahKO/cdf/KO/ko16.CDF"
## [3] "/usr/local/lib/R/site-library/faahKO/cdf/KO/ko18.CDF"
## [4] "/usr/local/lib/R/site-library/faahKO/cdf/KO/ko19.CDF"
## [5] "/usr/local/lib/R/site-library/faahKO/cdf/KO/ko21.CDF"
## [6] "/usr/local/lib/R/site-library/faahKO/cdf/KO/ko22.CDF"
## [7] "/usr/local/lib/R/site-library/faahKO/cdf/WT/wt15.CDF"
## [8] "/usr/local/lib/R/site-library/faahKO/cdf/WT/wt16.CDF"
## [9] "/usr/local/lib/R/site-library/faahKO/cdf/WT/wt18.CDF"
## [10] "/usr/local/lib/R/site-library/faahKO/cdf/WT/wt19.CDF"
## [11] "/usr/local/lib/R/site-library/faahKO/cdf/WT/wt21.CDF"
## [12] "/usr/local/lib/R/site-library/faahKO/cdf/WT/wt22.CDF"
raw_data <- readMSData(files = cdfs, mode="onDisk")- その他フォーマットのファイルの読み込み
- readMSData関数では,NetCDFの他に,mzml/mzXML/mzData形式のファイルを読み込むことが出来る
- 質量分析計から得られた測定データを読み込むには,msconvertなどを用いてcentroidのmzmlに変換して利用されることが多い
- 測定データを直接読み込むには,例えばThermo Fisher Scientific社の.rawファイルではBioconductorパッケージrawrrが利用できる.
ピークピッキング
データを読み込んだ後,ピークを拾い上げる処理であるピークピッキングを行う.ここでは,CentWaveと呼ばれるウェーブレット変換を用いた方法を用いている.
ピークピッキングのパラメーターはCentWaveParam関数で設定される.パラメーターの値は,XCMSのドキュメントに記載されたものをそのまま利用しているが,実際は試行錯誤で決定する必要がある
cwp <- CentWaveParam(peakwidth = c(20, 80), noise = 5000, prefilter = c(6, 5000))
xdata <- findChromPeaks(raw_data, param = cwp)アライメント
次に,クロマトグラムのアライメントと呼ばれる処理を行う.例として,保持時間が2760秒から2820秒までのtotal ion chromatogramを確認すると,以下に示すようにサンプル間で保持時間のズレが生じていることがわかる.
tic <- chromatogram(xdata, aggregationFun="sum")
plot(tic,xlim=c(2760,2820), peakType="none")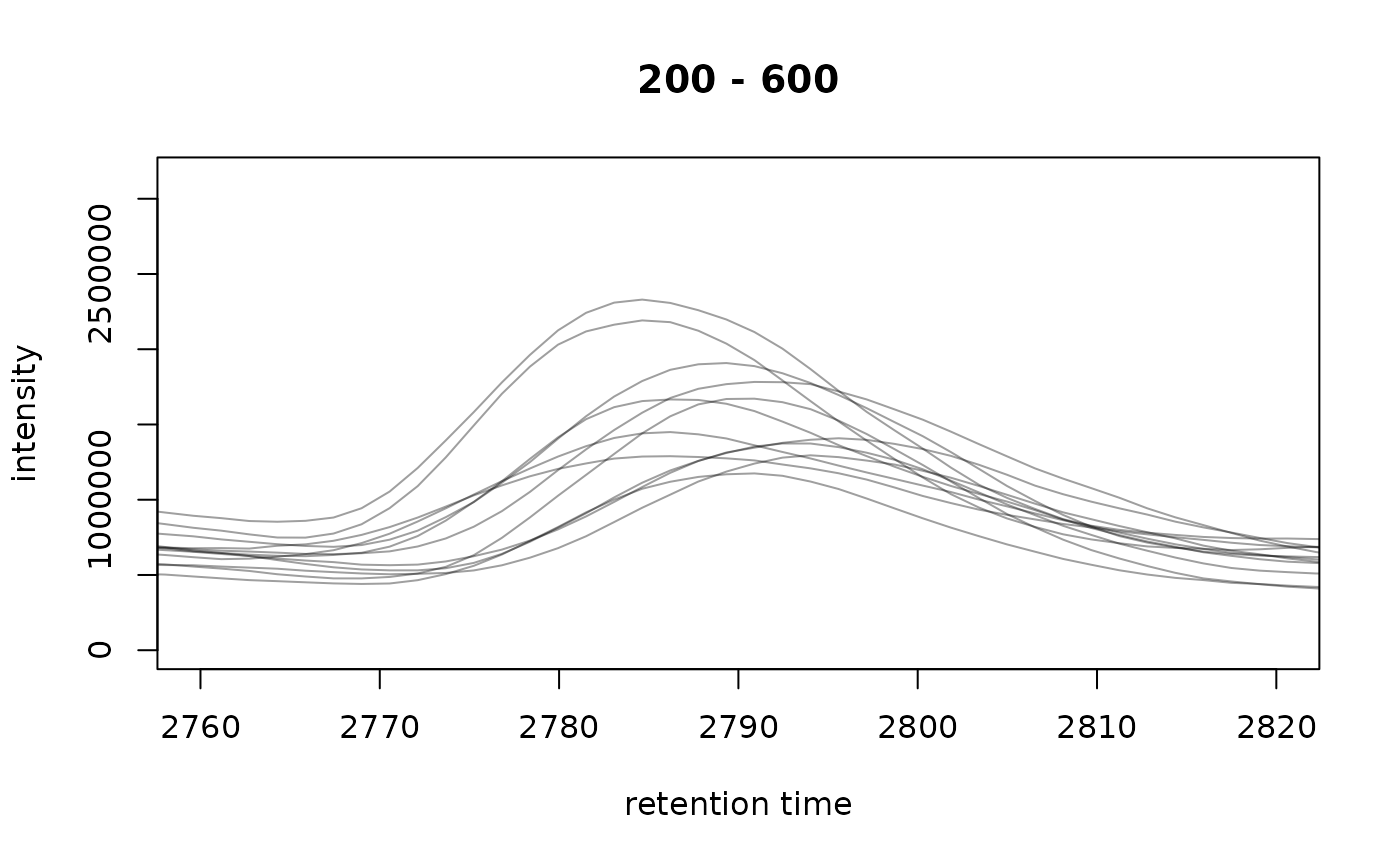
そこで,クロマトグラムの保持時間をObiwarp法を用いて補正した結果を次に示す.
xdata <- adjustRtime(xdata, param = ObiwarpParam(binSize = 0.6))
tic <- chromatogram(xdata, aggregationFun="sum")
plot(tic,xlim=c(2760,2820), peakType="none")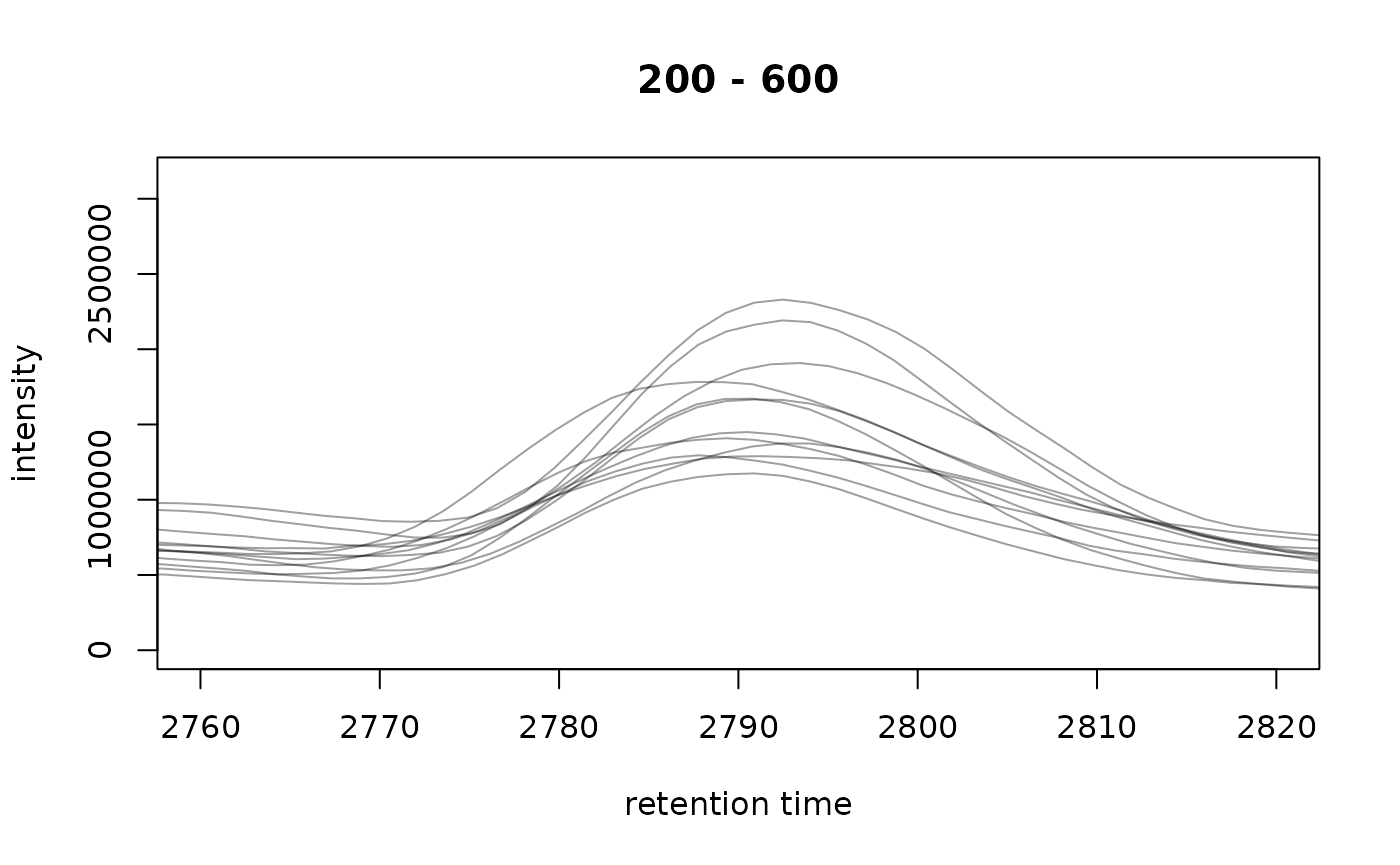
クロマトグラムのアライメントのパラメーターの値は,ピークピッキングと同様にXCMSのドキュメントに記載されたものを そのまま利用しているが,クロマトグラムの保持時間のズレ方は分析法によって異なることから,実際は試行錯誤により決める必要がある.
ピークの対応付け
次に,それぞれのサンプル間でのピークを対応付けをgroupChromPeaks関数を用いて行う. PeakDensityParamはこれまでと同様に,パラメーターを変えながら試行錯誤により決める必要がある.
## Group Peaks
sample_group <- c(1,1,1,1,1,1,1,1,1,1,1,1)
pdp <- PeakDensityParam(sampleGroups = sample_group, minFraction = 0.4, bw = 30)
xdata <- groupChromPeaks(xdata, param = pdp)sampleGroupsはここでは全て1のベクトルを設定したが,例えば以下
sample_group <- c(“KO”, “KO”, “KO”, “KO”, “KO”, “KO”, “WT”, “WT”, “WT”, “WT”, “WT”, “WT”)
のように,群毎に設定しても良い.
欠損ピークの穴埋め
ここまでの処理によって,各ピークとサンプルのデータ行列が作られ,次のようになっている.
head(featureValues(xdata))## ko15.CDF ko16.CDF ko18.CDF ko19.CDF ko21.CDF ko22.CDF wt15.CDF
## FT001 1924712.0 1757151.0 1714581.8 1220358.1 1383416.7 1180288.2 2129885.1
## FT002 213659.3 289500.7 194603.7 NA NA 178285.7 253825.6
## FT003 349011.5 451863.7 337473.3 NA 343897.8 208002.8 364609.8
## FT004 286221.4 NA 364299.5 NA 164009.0 149097.6 255697.7
## FT005 762727.3 NA 1345515.1 608016.5 380970.3 588986.4 1286883.0
## FT006 NA 235308.8 246563.6 NA NA NA 229292.1
## wt16.CDF wt18.CDF wt19.CDF wt21.CDF wt22.CDF
## FT001 1634342.0 1810112.28 1507943.1 1623589.2 1354004.9
## FT002 241844.4 228501.09 216393.1 240606.0 185399.5
## FT003 360908.9 54566.86 NA NA 221937.5
## FT004 311296.8 NA NA 366441.5 271128.0
## FT005 1739516.6 NA 214367.8 639755.3 508546.4
## FT006 135905.0 143917.91 NA NA NANAは欠損ピークである。欠損ピークは、再度測定データを確認して、実際にピークが存在するかどうか確認する必要がある。
xdata <- fillChromPeaks(xdata, param = ChromPeakAreaParam())
head(featureValues(xdata))## ko15.CDF ko16.CDF ko18.CDF ko19.CDF ko21.CDF ko22.CDF wt15.CDF
## FT001 1924712.0 1757151.0 1714581.8 1220358.1 1383416.7 1180288.24 2129885.1
## FT002 213659.3 289500.7 194603.7 154926.8 169833.5 178285.70 253825.6
## FT003 349011.5 451863.7 337473.3 107086.6 343897.8 208002.80 364609.8
## FT004 286221.4 293386.9 364299.5 210147.4 164009.0 149097.55 255697.7
## FT005 762727.3 885092.7 1345515.1 608016.5 380970.3 588986.38 1286883.0
## FT006 210804.9 235308.8 246563.6 103647.6 117760.3 71298.51 229292.1
## wt16.CDF wt18.CDF wt19.CDF wt21.CDF wt22.CDF
## FT001 1634342.0 1810112.28 1507943.1 1623589.2 1354004.93
## FT002 241844.4 228501.09 216393.1 240606.0 185399.47
## FT003 360908.9 54566.86 174070.9 232554.2 221937.53
## FT004 311296.8 255216.99 132769.2 366441.5 271128.02
## FT005 1739516.6 872221.89 214367.8 639755.3 508546.42
## FT006 135905.0 143917.91 118929.2 105907.5 85332.36fillChromPeaks関数により欠損ピークの穴埋めを行った後で、それでも欠損値になっていれば0を埋める
データ行列
data <- t(featureValues(xdata, value="into")) # sample*peak
data[is.na(data)] <- 0最後に主成分分析を行ってデータを可視化する。
